Каталитическая активность - комплекс
Cтраница 1
Каталитическая активность комплексов такого типа, согласно нашим данным, низка ( рис. 3); она значительно ниже активности даже наименее активных полихелатов. [1]
Каталитическая активность комплекса ЭДТА с Fe3 при избытке ЭДТА, исключающем образование каталитически активных биядерных комплексов [7], по-видимому, объясняется внешнесферным механизмом окисления-восстановления iFe3 в комплексе. [2]
Каталитическая активность винилпиридиновых комплексов МХЯ в сочетании с А1 ( цзо - С4Н9) 2С1 зависит от строения лиганда и уменьшается в ряду 4 - ВП 2 - СН3 - 5 - ВП 4 - СН3 - 5 - ВП 2 - ВП. Вероятно, этот ряд отражает как степень активации эвзо-циклической двойной связи в комплексносвязанном ВП, так и повышение устойчивости комплексов МХй-а ВП в отношении алкилирова-ния и обменного перекоординирования. [3]
Сравнение каталитической активности комплексов палладия, образующихся in situ из Pd ( OAc) 2 и Pd ( PPh3) 4 в реакции окислительного карбонилирования фенилацети-лена ( 1) с активностью комплексов III и IV показало, что все использованные соединения обладают соизмеримой каталитической активностью, что позволяет исключить из рассмотрения гипотезы, включающие соединения Pd ( II) в качестве активных частиц. [4]
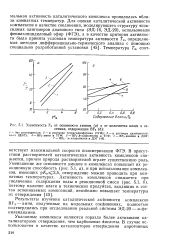 |
Зависимость Га от основности аминов ( а и от ( количества влаги в системах, содержащих BF3 ( б. [5] |
Результаты изучения каталитической активности комплексов BF3 - амин, полученные на модельных соединениях, полностью подтвердились при исследовании реальной системы ЭД-20 - м-фе-нилендиамин. [6]
Для оценки каталитической активности комплексов никеля ( и кобальта) как селективных катализаторов окисления этил-бензола в а-фенилэтилгидропероксид мы предложили использовать параметр 5 С; S - усредненная селективность окисления в ФЭГ, характеризующая изменение S в ходе окисления от значения S0 в начале реакции до некоторого Slim, условной величины, выбранной в качестве стандарта для серии сравниваемых каталитических систем. За Slim в данном случае принята glim § 00 / примерно равная селективности некатализированиого окисления этилбензола на начальных стадиях реакции. [7]
Предложена теория каталитической активности комплексов переходных металлов. Каталитическая активность определяется энергиями возбужденных состояний катализатора, которые имеют определенный спин и пространственную симметрию. Показано, что для эффективного катализа запрещенной по симметрии реакции изомеризация необходимо: I) наличие яизколэжащего возбужденного состояния катализатора со спином, отличающимся на единицу от спина основного состояния; 2) наличие достаточного числа состояний окисленной и восстановленной форм катализатора. Объяснено экспериментально наблюдаемое различие каталитической активности порфиринатов w, Ft, Мл в реакции изомеризации квадрициклана в норборнадиен. [8]
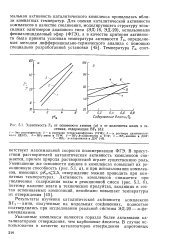 |
Зависимость Га от основности аминов ( а и от ( количества влаги в системах, содержащих BF3 ( б. [9] |
В присутствии растворителей каталитическая активность комплексов снижается, причем природа растворителей играет существенную роль. Уменьшение же основности аминов в комплексах повышает их реакционную способность ( рис. 5.1, а), и при использовании комплексов, имеющих р / ( а 2 5, отверждение можно проводить при комнатных температурах. [10]
Предварительные данные о каталитической активности комплексов описанного типа приведены в табл. 3, из которой следует, что закрепление на силикагеле приводит к получению стабильных катализаторов с высокой активностью. [11]
Для проверки гипотезы о каталитической активности комплексов Pd ( 0) в реакции ( 1) были синтезированы новые соединения ( [ Pd ( PPh3) 2QMH2Q), [ Pd ( PPh3) 2 ( Q) ] ( H2Q) 2Q, [ Pd ( PPhs) Q ] 2), структура которых была установлена методом РСА. Каталитическая активность этих соединений близка к активности системы, полученной из Pd ( OAc) 2, и-бензохинона и трифенилфосфина. [12]
Был установлен механизм контроля каталитической активности комплексов M ( L1) 2 ( М Ni, Co, L1 асас -) добавками электронодонорных монодентатных лигандов L2 ( L2 ГМФА, ДМФА, М - метилпирролидон-2) в реакции окисления этилбен-зола молекулярным кислородом. Под влиянием L2 в процессе окисления образуются два каталитически активных комплекса. На начальных стадиях реакции формирующиеся in situ первичные комплексы M ( L1) 2L2 тормозят образование фенола ( Ф), ответственного за дезактивацию катализатора. По мере развития процесса наблюдается увеличение 8ФЭг по сравнению с начальной стадией окисления и снижение w реакции. В развившейся реакции лиганды L2 контролируют трансформацию комплексов M ( L) 2 в более активные селективные частицы. При этом рост 8ФЭГ достигается за счет участия катализатора в реакции активации О2 и торможения цепного и ге-теролитического распада ФЭГ. Изменяется направление образования продуктов. [13]
Был установлен механизм контроля каталитической активности комплексов M ( L1) 2 ( М Ni, Co, L1 асас) добавками электронодонорных монодентатных лигандов L2 ( L2 ГМФА, ДМФА, М - метилпирролидон-2) в реакции окисления этилбен-зола молекулярным кислородом. Под влиянием L2 в процессе окисления образуются два каталитически активных комплекса. На начальных стадиях реакции формирующиеся in situ первичные комплексы M ( L1) 2L2 тормозят образование фенола ( Ф), ответственного за дезактивацию катализатора. По мере развития процесса наблюдается увеличение 8фЭг по сравнению с начальной стадией окисления и снижение w реакции. В развившейся реакции лиганды L2 контролируют трансформацию комплексов M ( L1) 2 в более активные селективные частицы. При этом рост 8ФЭГ достигается за счет участия катализатора в реакции активации О2 и торможения цепного и ге-теролитического распада ФЭГ. Изменяется направление образования продуктов. [14]
Был установлен механизм контроля каталитической активности комплексов M ( L1) 2 ( М Ni, Co, L1 асас -) добавками электронодонорных монодентатных лигандов L2 ( L2 ГМФА, ДМФА, М - метилпирролидон-2) в реакции окисления этилбен-зола молекулярным кислородом. Под влиянием L2 в процессе окисления образуются два каталитически активных комплекса. На начальных стадиях реакции формирующиеся ш situ первичные комплексы M ( L1) 2L2 тормозят образование фенола ( Ф), ответственного за дезактивацию катализатора. По мере развития процесса наблюдается увеличение 8ФЭГ по сравнению с начальной стадией окисления и снижение w реакции. В развившейся реакции лиганды L2 контролируют трансформацию комплексов M ( lJ) 2 в более активные селективные частицы. При этом рост 8ФЭг достигается за счет участия катализатора в реакции активации О2 и торможения цепного и ге-теролитического распада ФЭГ. Изменяется направление образования продуктов. [15]
Страницы: 1 2 3