Измерение - начальная скорость - реакция
Cтраница 1
Измерение начальных скоростей реакции является особенно полезным для получения констант скоростей медленных реакций. [1]
Измерение начальной скорости реакции ( dx / dt) 0 - - r представляет собой еще один метод определения порядка реакции. [2]
Для измерения начальной скорости реакции были разработаны методы автоматического и ручного манипулирования. [3]
 |
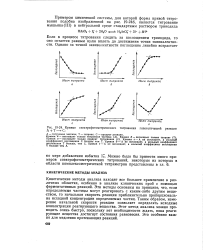
Калибровочные кривые для систем, подчиняющихся закономерности. начальная еюсь рость Л [ С ] ис1 5 ( текст. [4] |
Использование подхода, основанного на измерении начальной скорости реакции, имеет много преимуществ. Для оценки начальной скорости реакции применяют два основных метода, которые позволяют обойтись без построения всей кинетической кривой. В методе фиксированной концентрации измеряют время, необходимое для достижения строго заданного состава реакционной смеси. В методе фиксированного времени определяют изменение состава смеси Дя за строго определенный промежуток времени. [5]
Методы анализа, основанные на измерении начальной скорости реакций, имеют ряд значительных преимуществ. Поскольку количество продукта, образующегося за период измерения, невелико, обратная реакция не уменьшает общую результирующую скорость в заметной степени. Осложнения, вызываемые более медленными побочными реакциями, обычно минимальны. Поскольку концентрации реагентов в ходе реакции почти не изменяются, реакция имеет псевдонулевой порядок. Для реакций, скорости которых лежат в приемлемых пределах, измерения начальной скорости, по-видимому, должны быть более точными, чем измерения при больших глубинах протекания реакции, поскольку наивысшая скорость наблюдается вначале. При этом наклон кривой максимально крутой, а соотношение сигнал / шум наиболее благоприятно. [6]
Зависимость констант скорости, полученных при измерении начальных скоростей реакции, от концентрации различных реагентов легко можно изучить в экспериментах, в которых раздельно варьируются концентрации этих реагентов. Небольшие количества высокореакционноспособных примесей в реагирующих веществах также могут привести к ошибкам в кинетических измерениях по методу начальных скоростей, как и по методу псевдопервого порядка. [7]
При проведении экспериментов с разбавленным водородом возможно измерение только начальных скоростей реакции, когда объем газа в газовой бюретке уменьшается не более чем па 10 % и изменением состава газа из-за поглощения водорода можно пренебречь. [8]
Для определения порядка в данном случае используют метод Вант-Гоффа, называемый методом исключений или методом измерения начальных скоростей реакций. Принцип метода заключается в следующем. [9]
Исследована кинетика ферментативного гидролиза ацетилхолина, ацетил-р-метил-холина, бутирилхолина и бензоилхолина, катализируемого холин-эстеразой сыворотки крови лошади ( очищенный препарат фермента с уд. Для измерения начальной скорости реакции использован потенциометрический метод с регистрирующим рН - метром. [10]
Научные исследования относятся преимущественно к органической химии. Впервые применил ( 1866) органические соединения для определения строения неорганических соединений: решая вопрос о строении фосфористой кислоты, использовал ее органические производные. Посредством измерения начальных скоростей реакций открыл ( 1877 - 1897) закономерности, устанавливающие влияние строения спиртов и органических кислот на - корость и предел этерифи-кации Показал, что эти результаты применимы в качестве критериев разграничения изомерных первичных, вторичных и третичных спиртов Посредством определения констант скоростей реакций установил влияние природы растворителя ( 1886 - 1889) и температуры ( 1889) на процессы образования и разложения аминов и амидов кислот. Нашел ( 1882), что продукты реакции оказывают влияние на процесс термического разложения третичного амилацетата. [11]
 |
Кривые спектрофотометрического титрования А Т - С. [12] |
Кинетические методы анализа находят все большее применение в различных областях, особенно в анализе клинических проб с помощью ферментативных реакций. Эти методы основаны на принципе, что, если определяемые частицы могут реагировать с каким-либо другим веществом, то начальная скорость реакции приблизительно пропорциональна исходной концентрации определяемых частиц. Таким образом, измерение начальной скорости реакции позволяет определять исходные концентрации реагирующего вещества. Этот метод анализа можно проводить очень быстро, поскольку нет необходимости ждать, пока реагирующие вещества достигнут состояния равновесия. Это особенно важно для медленно протекающих реакций. [13]
Существует ряд примеров, свидетельствующих как будто о том, что ферменты катализируют реакцию более эффективно в одном направлении, чем в другом. Однако это никоим образом не противоречит принципу микроскопической обратимости, поскольку условия, в которых проводили измерения, различаются для двух направлений реакции; в условиях равновесия фермент должен осуществлять равный катализ в обоих направлениях. Чтобы удовлетворять соотношению Хелдена, отношение констант скоростей kf / kr, которое отлично от Ketl, должно согласовываться с величинами Kg и Кр. Так, если катализ реакции в прямом направлении много больше, чем в обратном ( в условиях, в которых фермент насыщен субстратом или продуктом при измерении начальных скоростей реакции в каждом направлении), константа диссоциации Kg должна быть больше, чем Кр. Такие малые значения Кр часто приводят к сильному ингибированию продуктом реакции в прямом направлении. [14]
Однако при этом предполагается, что изучаемая реакция не осложнена наличием обратной реакции. В данном случае окисление уксусного альдегида шестивалентным хромом протекает практически необратимо. Для нахождения энергии активации строится график зависимости логарифма обратной величины времени, в течение которого прореагирует данный строго выбранный объем окислителя при разных температурах, от обратной температуры. На рис. 13 представлена эта зависимость. Найденное значение энергии активации равно 24 0 ккал / М и находится в хорошем согласии с величиной, полученной по измерениям начальной скорости реакции. [15]
Страницы: 1