Ход - экспериментальная кривая
Cтраница 2
Разумеется, это согласие не свидетельствует о широкой применимости предложенной модели, и в каждом конкретном случае необходим самостоятельный анализ факторов, определяющих ход экспериментальных кривых. [17]
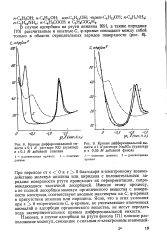
При увеличении количества смеси, вводимой в колонку, ВЭТТ, начиная с некоторого значения температуры, резко возрастает при повышении рабочей температуры колонки. Такой ход экспериментальных кривых теоретически может быть объяснен влиянием на эффективность препаративной колонки степени разбавления Кт и таких членов уравнения ( 5), как ( 3 и у. Из полученных в ходе исследования данных следует, что величина Кт с ростом температуры вначале резко убывает, а затем, начиная с 70 - 75, остается постоянной. Эта температура близка к средней температуре кипения компонентов разделяемой смеси. [18]
Ранее [79] мы рассмотрели схему самовоспламенения водородо - кисло-родной смеси в ударных волнах и показали, что по опытным данным о зависимости задержек воспламенения от температуры можно выявить некоторые характерные особенности механизма процесса - существование ведущей реакции в области высоких температур и появление ( при переходе к более низкой температуре) предела, связанного с обрывом разветвленных цепей в объеме. Существующие сейчас опытные данные по задержкам воспламенения в обла-сти температур от 800 до 2800 К довольно разноречивы. Так, по данным работы [81], в метано-воздушных смесях существует излом в ходе экспериментальной кривой в координатах Igt-1 / 7 при Т - 1600 К, соответствующий переходу от энергии активации 8 26 ккал / моль при более высоких температурах к е 85 ккал / моль в низкотемпературной области. В работе [82], наоборот, наблюдался переход от е - 53 ккал / моль к е - 20 ккал / моль соответственно приблизительно в той же температурной области. В работе [83] для интервала 1800 - 2500 К энергия активации оценивается величиной около 33 ккал / моль. Наконец, в недавней работе [84] было также получено е 33 8 ккал / моль при 1050 - - 2100 К ДЛЯ, периода индукции реакции в целом и к 21 5 ккал / моль для задержки появления гидроксила. Анализ полученных данных ограничивается обычно сопоставлением с низкотемпературным механизмом. Лишь в работе [83] сделана попытка ввести разветвление через Н О2 - ОН О ( е 16 - 18 ккал / моль), что не соответствует найденной там же энергии активации процесса. [19]
 |
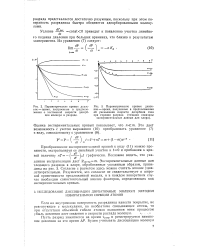
Кривые дифференциальной емкости в 0 1 Л. растворе КС1 ( пунктир и с 0 1 М. добавкой анилина.| Кривые дифференциальной емкости в 1 N растворе NaaSOi ( пунктир и с 0 05 УИ добавкой фенола. [20] |
При переходе отв 0к80 благодаря я-электронному взаимодействию молекул анилина или пиридина с положительными зарядами поверхности ртути происходит их переориентация, сопровождающаяся частичной десорбцией. Именно этому процессу, а не полной десорбции молекул органического вещества с поверхности электрода, соответствуют анодные максимумы на С, ф-кривых в присутствии анилина или пиридина. Ясно, что в этих условиях при е 0 теоретические С, ф-кривые, не учитывающие отмеченных аномалий в адсорбции органического вещества, не могут передать хода экспериментальных кривых дифференциальной емкости. [21]
Можно ожидать, что молекулярные веса полимеров, имеющих не очень высокий начальный молекулярный вес, будут несколько уменьшаться уже в самом начале реакции. Однако эти очень малые изменения трудно обнаружить, что осложняется еще и широким распределением по молекулярным весам, характерным для этих полимеров. Так, например, полимер, имеющий молекулярный вес 179 000, содержит определенное количество небольших молекул, способных к полной деструкции. Поэтому ход экспериментальной кривой определяется двумя факторами: с одной стороны, удаление малых молекул приводит к увеличению молекулярного веса, с другой-частичная деструкция больших молекул уменьшает его. [22]
Результаты расчета приведены в табл. 5.1. Видно, что при длительности релаксационного процесса 180 мин экспериментальные кривые описываются пятью временами релаксации. Остальные времена релаксации качественно согласуются с найденными зависимостями (5.80), хотя наблюдается значительное количественное расхождение. Это объясняется принятыми при выводе этих формул допущениями и упрощением исходных дифференциальных уравнений. Таким образом, полученное решение показывает, что предложенная модель правильно передает ход экспериментальных кривых и позволяет объяснить закономерное появление спектра времен релаксации. На самом деле поведение системы может характеризоваться двумя основными временами релаксации. Остальные времена являются комбинацией этих двух основных времен и зависят от деформации и упругих характеристик полимера. [23]
 |
Времена релаксации для образца блок-сополимера полистирола. [24] |
Для экспериментальной проверки полученных соотношений были рассчитаны спектры вр. Результаты расчета приведены в табл. 5.1. Видно, что при длительности релаксационного процесса 180 мин экспериментальные кривые описываются пятью временами релаксации. При этом времена TI и t2 практически не зависят от деформации & и составляют в среднем ti 12 3 с и ( т2 1.34 - 103 с. Остальные времена релаксации качественно согласуются с найденными зависимостями (5.80), хотя наблюдается значительное количественное расхождение. Это объясняется принятыми при выводе этих формул допущениями и упрощением исходных дифференциальных уравнений. Таким образом, полученное решение показывает, что предложенная модель правильно передает ход экспериментальных кривых и позволяет объяснить закономерное появление спектра времен релаксации. На самом деле поведение системы может характеризоваться двумя основными временами релаксации. Остальные времена являются комбинацией этих двух основных времен и зависят от деформации и упругих характеристик полимера. [25]
Некоторые молекулярные структуры, установленные ранее визуальным методом, оказались ненадежными или даже вовсе неверными, в результате чего метод дифракции электронов незаслуженно приобрел плохую репутацию. Однако, как мы увидим в дальнейшем, прогресс в технике и в методике интерпретации позволяет при определенных условиях получать очень точные значения молекулярных параметров. Существуют две основные причины допущенных ранее ошибок. Во-первых, рассеяние, обусловленное легкими атомами, в особенности водородом, невелико, и оно может быть замаскировано рассеянием на более тяжелых атомах. Во-вторых, число дифракционных колец редко превышает десять, так что для определения параметров молекулы имеется не более двадцати максимумов и минимумов. Для некоторых молекул этого оказывается недостаточно и иногда казалось, что совершенно неправильные модели удовлетворительно передают ход экспериментальной кривой. [26]
Страницы: 1 2